FAQ: Frequently Asked Questions
- I would like to get access to the HPC facilities at ISU. How do I do that, and how much will it cost?
- I need help using clusters. Who do I contact?
- I bought a new phone or erased GA. How can I reset GA?
- What is ssh and scp? Where do I find them?
- I get "WARNING: REMOTE HOST IDENTIFICATION HAS CHANGED!" message. What should I do?
- I don't like having to authenticate so often. Is there a way to login less?
- FileZilla does not ask me for verification code. What do I do to connect to my account on a cluster which uses Google Authenticator?
- What is the queue structure?
- I want one of my jobs to run before the others. What do I do?
- Why is my job waiting in the queue while there are nodes available?
- I get "Disk quota exceeded" error message but I don't have a lot of files in my home directory. What can I do?
- I need a specific software package to be installed. What do I do?
- How can I use Matlab on a cluster?
- How can I use R on a cluster?
- I need an R package to be installed. What do I do?
- I need to use Python on a cluster?
- I need a Python package that isn't installed, what do I do?
- I need a different version of Python than what is available via modules. How do I get it?
- I need a Perl package that isn't installed, what do I do?
- How can I use ASREML on a cluster?
- How can I move files between the cluster and my personal computer or another cluster?
- How can I access MyFiles from the clusters?
- My compile or make commands fail with a segmentation fault. What should I do?
- How do I make specific modules be automatically loaded on the compute nodes assigned to my job?
- I get "module: command not found" error when trying to run my job. Are modules installed on the clusters?
- What to do when the job runtime exceeds max queue time?
- How to debug my program?
- I'm a Windows/Mac user. How to use Unix?
- I get "Disk quota exceeded" error message when trying to remove files. What can I do?
- My ssh session dies with the messages "Write failed: Broken pipe" after a few minutes of inactivity. How do I fix this?
- New to Slurm? Here are some useful commands.
- How do I compile software for use on the cluster?
- How can I see html files on a cluster?
- I dont want my jobs to requeue on failure, how do I stop that?
- How do I use GNU Parallel to maximize my usage of a node?
I would like to get access to the HPC facilities at ISU. How do I do that, and how much will it cost?
One can not buy compute time on the clusters listed at http://www.hpc.iastate.edu/systems.
The Nova cluster has three tiers – sponsored, educational and free. The sponsored portion of the Nova cluster can be used only by those who purchased nodes and storage on the Nova cluster. These researchers can either directly add users to their allocation at https://asw.iastate.edu/cgi-bin/acropolis/list/members or request access for their students and colleagues by sending an email to hpc-help@iastate.edu .
Faculty can request access for them and their students to the free and educational Nova tiers filling out this form .
If your group would like to purchase nodes on the Nova Cluster, submit the Nova purchase form. College of LAS has purchased nodes and storage for their researchers. To obtain an account, contact researchit@iastate.edu .
I need help using clusters. Who do I contact?
If you need help please send an email to hpc-help@iastate.edu . In the email specify the name of the cluster. When sending email from non-iastate email address, also specify your ISU NetID. If applicable add the following information:
- The command you ran (sbatch ...)
- The directory you ran it from
- The job ID
- Any output you received
- Any additional information that would help us help you.
I bought a new phone or erased GA. How can I reset GA?
If you logged your phone number in the system, you can reset your GA at https://hpc-ga1.its.iastate.edu/reset/ . Note that you need to be on campus or use ISU VPN in order to access this page. To log your phone number where you can receive text message, issue phone-collect.sh on the nova login nodes.
What is ssh and scp? Where do I find them?
ssh is a program for logging into a remote machine and for executing commands on a remote machine. To be able to use our clusters you will need to ssh to them. If your computer is running Linux or OSX you already have ssh command (open terminal and type “ssh <name_of_the_cluster>”). If your login name on your computer is different from the login name on the cluster (which is the same as University NetID), add your NetID to the ssh command: “ssh <NetID>@<name_of_the_cluster>” or “ssh –l <NetID> <name_of_the_cluster>”. scp command is used to copy files between different machines on a network. It's also available on the Linux and Mac computers.
If you’re using Windows computer, you probably don’t have ssh and scp available. In that case you can download free SSH client PuTTY and free SFTP/SCP client WinSCP. Some people use SFTP client FileZilla which is also available for MacOS. Both WinSCP and FileZilla have graphic interface. We recommend using Globus Connect to transfer files to/from ISU HPC Clusters.
I get "WARNING: REMOTE HOST IDENTIFICATION HAS CHANGED!" message. What should I do?
You will receive such a message if the host key for the host you're trying to connect to (e.g., novadtn.its.iastate.edu) has changed since the last time you tried to access this host. The message will say in which line of your known_hosts files it found an old host key.
To get rid of this message, issue the following command on your computer from which you're trying to connect to the host:
ssh-keygen -R <hostname>
Replace <hostname> with the right name for the host, e.g., novadtn.its.iastate.edu. This will remove all keys belonging to novadtn.its.iastate.edu from your known_hosts file.
I don't like having to authenticate so often. Is there a way to login less?
If you using Windows cmdline tools, any Linux, or MacOS you can use ssh's "ControlMaster" option so all of your connections after the first one go though the first one so additional authentications are not required. This allows you to have multiple shells open while only authenticating once to a given cluster. This works for for ssh, scp, and sftp. To set this up:
- go into your home directory
- then into your .ssh folder
- edit or create a file called "config"
- Using the Nova cluster as an example the contents of the file should be:
Host nova.its.iastate.edu
IdentityFile ~/.ssh/id_rsa
ControlMaster auto
ControlPath ~/.ssh/%r@%h:%p
ControlPersist yes
ServerAliveInterval 60
Of course substitute the name of the cluster you are using on the "Host" line if your using a different cluster. You can have multiple blocks like this in the config file if you use multiple clusters.
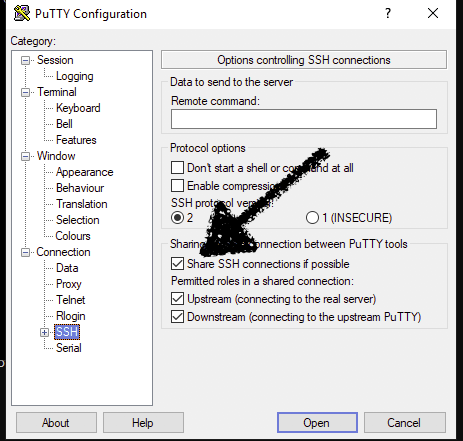
On a windows box using putty this can be done by checking the "Share SSH connections if possible" button in the SSH section of the putty config dialogs.
We also suggest you try the "tmux" command.
Tmux will allow you to have many shells in a single window all with a single authentication. If your VPN or WiFi connection drops you will only have to login once and then reattach to your sessions with "tmux attach" This also has the advantage that anything you were running at the time stays running as well.
Quick tips to get started, just type: tmux
This starts tmux and you are in shell "0"
To start a second shell use control-b c (^b c) this will open a second shell "1." You can open as many as you wish in this manner.
To switch between them use "^b #" where # is the number of the shell you want to switch to as seen on the bar on the bottom.
To exit the shells just close them with "exit" or "^d" at a shell prompt when you are finished with them.
Use "man tmux" for more information on using tmux.
FileZilla does not ask me for verification code. What do I do to connect to my account on a cluster which uses Google Authenticator?
In FileZilla click on the “File” menu section and choose “Site Manager”. In the Site Manager window click on “New Site”, enter Host (e.g.novadtn.its.iastate.edu), set Protocol to SFTP, set Logon Type to Interactive, type your NetID in the User field. You will also want to open the "Transfer Settings" tab and check "Limit number of simultaneous connections." Set the "maximum number of connections" to "1" If you do not do this you will be prompted for your verification code and password many times. Click on Connect and a small window will open, showing message from the cluster prompting for Verification code. In the Password field enter the 6 digit number generated by the GA running on your mobile device. Next you will be prompted for password.
What is the queue structure?
When you submit a job with sbatch/salloc/srun command it gets into routing queue which places the job into appropriate queue based on the number of nodes and wall time requested. To see the current queue structure on a specific cluster, issue 'sinfo'. To see the jobs in the queue, issue 'squeue'.
I want one of my jobs to run before the others. What do I do?
Issuing:
scontrol update dependency=after:113271 jobid=111433
and
scontrol update dependency=after:113271 jobid=111434
will create a dependency so that neither job 111433 nor 111434 will run until after 113271 completes.
Assuming that you do not submit any more jobs, this will ensure that job #113271 will be the next job of yours that runs.
Why is my job waiting in the queue while there are nodes available?
There might be two reasons:
1. A downtime is scheduled. If a job won't be able to complete before downtime, it will be waiting in the queue.
2. There are other jobs of higher priority waiting to obtain enough nodes to run .
You can issue:
squeue -a --start
to see estimated start dates assuming new jobs with higher priority are not submitted, and assuming that jobs run to the wall time given on submission.
I get "Disk quota exceeded" error message but I don't have a lot of files in my home directory. What can I do?
Some software creates hidden directories in the home directory. To see data usage in those directories, issue:
du -sh ~/.??*
Often those would be .conda or .singularity . Those directories can be moved to the group working directory and symbolic links can be created in the home directory to point to the directories moved to /work/<group_name> . For example:
cd
mv .singularity /work/<group_name>/$USER
ln -s /work/<group_name>/$USER/.singularity .singularity
Files in .cache may also contribute to your home directory usage. Those files can be safely deleted.
I need a specific software package to be installed. What do I do?
First of all check whether this software is already installed and can be loaded as a module. For this issue the 'module avail' command. To load a specific module issue 'module load <module_name>'. You can also unload, swap and purge modules. See the output of 'module --help' or 'man module' for additional information.
The group specific software should be installed in the group working directories where all members of the group have access. Most software packages don't need superuser privileges to be installed. These are generic instructions that work in many cases:
- use 'wget URL_TO_TAR_FILE' to download source code (e.g. wget https://julialang.s3.amazonaws.com/bin/linux/x64/0.4/julia-0.4.5-linux-x86_64.tar.gz)
- use 'tar xvzf your_tar_file' to unpack *.tar.gz file that you downloaded, usually a new directory will appear in the current directory
- cd to the unpacked directory, and run './configure --prefix=/your_work_directory' (software will be installed in this directory)
- issue 'make' to compile software
- issue 'make install' to install compiled code in the directory specified in the --prefix option on the configure command
If you need help installing software send email to hpc-help@iastate.edu.
If you're part of LAS, you may send request to researchit@iastate.edu, and the LAS IT staff may install your software package as a module.
How can I use Matlab on a cluster?
Matlab is installed on all clusters. To use it you need to load matlab module. Since we don’t have the Distributed Compute Manager license, users can run Matlab only on one node, and not across multiple nodes. Remember to NOT run Matlab on the head node. Instead generate job script using the scriptwriter for the cluster where you want to run Matlab (see appropriate User Guide at http://www.hpc.iastate.edu/guides). In the script add the following commands:
module load matlab/R2018a
matlab -nodisplay -nosplash -nodesktop < program.m > matlab_output.log
Replace program.m with the name of your Matlab program. The output of the matlab command will be redirected to file matlab_output.log . If a different version of Matlab is needed, replace matlab-R2015a with the appropriate matlab module name.
How can I use R on a cluster?
To use R that is installed on the clusters you may need to load R module. Remember to NOT run R on the head node. Instead generate job script using the scriptwriter for the cluster where you want to run R (see appropriate User Guide at http://www.hpc.iastate.edu/guides). In the script add the following commands:
module load r
Rscript program.R
Replace program.R with the name of your R program. The "module load" command above will set environment for the default version of R. If a different version of R is needed, replace "r" with the appropriate R module name. To see available modules issue "module spider r".
I need an R package to be installed. What do I do?
Before installing an R package check if it's available as a module:
module avail <lower_case_name_of_the_package>
or
module spider <lower_case_name_of_the_package>
Most of the R packages can be installed in the home directory which is mounted on all nodes of a cluster. To use R that is installed on the clusters you may need to load R module:
module load r
To run your R programs follow the instructions at How can I use R on a cluster? and remember to NOT run R on the head node. However to install an R package, issue R command on the head node:
R
Within R issue the following command:
install.packages("name_of_the_package", repos="http://cran.r-project.org")
(replace name_of_the_package in the command above with the name of your package). It will issue a warning about "/shared/software/LAS/R/3.2.3/lib64"' not being writable and ask whether you would like to use a personal library instead. Reply "y" to this and to the following question on whether you would like to create a personal library ~/R/x86_64-pc-linux-gnu-library/3.2 .
If you want to use another location rather than the default location, for example, ~/local/R_libs/, you need to create the directory first:
mkdir -p ~/local/R_libs
Then type the following command inside R:
install.packages("name_of_the_package", repos="http://cran.r-project.org", lib="~/local/R_libs/")
To use libraries installed in this non-default location, create a file .Renviron in your home directory, and add the following line to the file:
export R_LIBS=~/local/R_libs/
To see the directories where R searches for libraries, use the following R command:
.libPaths();
If during install you see error messages that mention C99 mode or f77 compiler not being found, load gcc module before starting R:
module load gcc
I need to use Python on a cluster
Each cluster has an least one and usually several versions of python installed. To see which ones are available use:
module avail python
Then load the module for the version you want to use. If you don't need a particular version often
module load python
is enough. This will get you the most recent 3.X.Y Python has almost no modules installed by default you will need to load the ones you need. To see the list of available modules use:
module avail py-
python3 module will contail -py3- and python2 modules will contain -py-
I need a Python package that isn't installed, what do I do?
You will need to make a Python Virtual Environment or Conda Environment to install the package in.
I need a different version of Python than what is available via modules. How do I get it?
You can create a conda/mamba environment for any version of python.
For example, to install python 2.7 in the group working directory issue:
cd /work/<path_to_your_directory> module load micromamba export MAMBA_ROOT_PREFIX=/work/<path_to_your_directory> micromamba env create -n py27 python=2.7 -c conda-forge
To use it:
module load micromamba eval "$(micromamba shell hook --shell=bash)" export MAMBA_ROOT_PREFIX=/work/<path_to_your_directory> micromamba activate py27
I need a Perl package that isn't installed, what do I do?
There are multiple ways to install modules in Perl and depending on the use case, one may be preferable over the others.
cpanm is a convenient tool to install modules in the home directory.
$ module load perl
$ cpanm Test::More
Running the above command creates a “perl5” directory within the users’ home directory and contains all the required binaries and libraries associated with the “Test::More” module. Add PERL5LIB to the environment
$ export PERL5LIB=$HOME/perl5/lib/perl5
Perl modules can also be installed in other directories. For example –
the user can have the modules available to the rest of the project members so that everyone involved works in the same environment.
local::lib provides the flexibility to users to install modules in any custom
location(as long as they have write permissions).
$ eval "$(perl -I/path/to/the/project/dir/perl5/lib/perl5 -
Mlocal::lib=/path/to/the/project/dir/perl5)"In addition to creating “perl5” directory within the specified project directory, the above
command adds perl local::lib environment variables.
Then use cpanm to install the required modules.
$ cpanm Test::More
Note: The package source files will still be downloaded to your home directory
(~/.cpanm/sources)
Note: All users working on the project can access modules that were installed using local:lib, but only the original user can perform perl module installs.
How can I use ASREML on a cluster?
Load asreml module and run command "asreml". This command will either launch R (when module asreml/3.0 is loaded) or the stand-alone asreml command prompt (when module asreml/4.1 is loaded). If you load module asreml/3.0 , after issuing "asreml" command, at the R prompt type "library(asreml)".
How can I move files between the cluster and my personal computer or another cluster?
There are several ways to transfer files. The user guides for specific clusters (see http://www.hpc.iastate.edu/guides) in section "How to logon and transfer files" describe how to use scp. Data transfer node (novadtn) should be used to transfer large and multiple files. Remember to copy large amounts of data directly to your group working directory, and not to home directory.
How can I access MyFiles from the clusters?
MyFiles is accessible from login and data transfer node (novadtn). If it's been a while since you last authenticated to the cluster, issue 'kinit' and enter your university password. After entering your password, you will be able to cd to /myfiles/Users/$USER and to /myfiles/<deptshare>, where <deptshare> is your departmental share name; this is usually your department or college’s shortname, such as engr or las.
My compile or make commands on the head node fail with a segmentation fault. What should I do?
It is possible that the system resource limits on the head node are too strict to compile your programs. Try to either get an interactive session on a compute node or login to the data transfer node, novadtn, and issue your commands there. If that does not help, send an email to hpc-help@iastate.edu .
How do I make specific modules be automatically loaded on the compute nodes assigned to my job?
There are several ways to do this. You can include module commands in your job script. If you would like a specific set of modules to be loaded every time you login to a cluster, as well as on the compute nodes, add the necessary module commands to the .bashrc file in your home directory. If you have the necessary modules loaded and environment variables set, using -V option on the qsub command will export all the environment to the context of batch job.
I get "module: command not found" error when trying to run my job. Are modules installed on the clusters?
Yes, modules are installed on all clusters. If you see the list of available modules when issuing 'module avail' on the head node, but the 'module load' command in the job script fails with the message "module: command not found", it might be that you're not a bash user. To see the current shell, issue 'echo $0'. If your default shell is tcsh or csh, you can either change it to bash or replace' #!/bin/bash' in your job script with '#!/bin/csh'. To change default shell for hpc-class, go to https://asw.iastate.edu/cgi-bin/acropolis/user/shell.
What do I do when the job runtime exceeds max queue time?
Option 1: Get the answers faster:
- Use the fastest library routines. E.g. if dense linear routines are used in the code to solve systems of linear equations, a large increase in speed may be possible by linking with the vendor supplied routines. Link with the MKL library rather than non-optimized libraries.
- Change to a more efficient algorithm. This is the best since you get your answers quicker. HPC group can help you with numerical aspects and some algorithm choices, but you would need to supply the modeling knowledge.
- Go parallel. The program can be rewritten to use MPI. This often takes a long time but usually gives the best performance. The program can also be modified with OpenMP directives to perform portions of the program in parallel, and compiled to use all cores in a single node. The speedup is limited to the number of cores, and if not done well can even slow down a program.
Option 2: Use checkpointing.
Major production codes are checkpointed. In checkpointing, you periodically save the state of the program in a restart file. Whenever you run your program it reads the restart file to pick up from the last checkpoint. The advantage of this is that there is no limit to the total amount of time you can use. Barring disk crashes or total loss of the machine, your total runtime is indefinite, you just keep submitting the same job and start from where you left off. There is overhead associated with each checkpoint, and time executed after the last checkpoint is lost whenever the job is stopped.
How to debug my program?
Programs can appear to have no bugs with some sets of data because all paths through the program may not be executed. To debug your program, it is important to try and test your program with a variety of different data sets so that (hopefully) all paths in your program can be tested for errors.
Some bugs can be found at compile and/or run time when using compiler debugging options. We suggest to use the following debugging options for Intel compilers. (These compilers are available on all clusters.)
The “-g” option produces symbolic debug information that allows one to use a debugger. The “-debug” option turns on many of the debugging capabilities of the compiler. The “-check all” option turns on all run-time error checking for Fortran. The “-check-pointers=rw” option enables checking of all indirect accesses through pointers and all array accesses for Intel’s C/C++ compiler but not for Fortran. The “-check-uninit” option enables uninitialized variable checking for Intel’s C/C++ compiler (this functionality is included in Intel’s Fortran “-check all” option). The “-traceback” option causes the compiler to generate extra information in the object file to provide source code trace back information when a severe error occurs at run-time.
Intel’s MPI Checker provides special checking for MPI programs. To use MPI Checker add the “-check-mpi” option to the mpirun command.
Note that using options above will likely slow the program, so these options are not normally used when not debugging.
You can also use a debugger to debug a program. On the Nova cluster we recommend using Allinea’s DDT debugger. See instructions on how to use DDT in the Guides section on the left. The following debuggers are also available: Intel’s Inspector XE (inspxe-cl and inspxe-gui), GNU’s gdb and PGI’s pghpf.
When program does not complete and coredumpsize is set to a non-zero value one or more core files may be produced and placed in the working directory. If the limit on coredumpsize is not large enough to hold the program's memory image, the core file produced will not be usable. To change the configuration to allow useful core files to be produced enter “unlimit coredumpsize” or “ulimit –c unlimited”. Then a debugger can be used to analyze the core file(s).
I'm a Windows/Mac user. How to use Unix?
There are several ways to learn how to use Unix. You can take Math/ComS/CprE 424X class (Introduction to High Performance Computing) or a workshop organized by Genome Informatics Facility. You can take an online class or just search on Internet. For a quick introduction to Unix try the following Tutorial. You may also find useful the following materials: Slides from Workshop: Basic UNIX for Biologists.
I get "Disk quota exceeded" error message when trying to remove files. What can I do?
It looks like the disc is 100% full. Try to replace one of the files that you want to remove with an empty file by issuing the following command: "echo > file", where file is the name of your file. This command will write end-of-file into the file, and its size will become 1. Now the disc has some room, and you should be able to remove all the files that you want without getting "Disk quota exceeded" error message.
My ssh sessions dies with the messages "Write failed: Broken pipe" after a few minutes of inactivity. How do I fix this?
If your logging in from a Mac or Linux machine create a a directory called .ssh in your home directory. In that directory create a file called config with these contents:
Host *
ServerAliveInterval 60
Run "chmod 0600 config" on the file after creating it or it wont work and the system will complain.
If you are using putty in windows look in the Putty configuration for your host. Under Connection you will see "Seconds between Keepalives" set this to 60.
New to Slurm? Here are some useful commands.
This page has a summary of some many useful slurm commands: https://rc.fas.harvard.edu/resources/documentation/convenient-slurm-commands/
How do I compile software for use on the cluster?
First, on the head node I used salloc to get access to compute node to build the code ( this isn't necessary, but I try to avoid stressing out the head node):
$ salloc -N 1 --ntasks=4 --time 1:00:00
Then I create a work directory for this:
mkdir /work/ccresearch/jedicker/fftw
(obviously, your path will be different)
$ cd /work/ccresearch/jedicker/fftw
$ wget http://www.fftw.org/fftw-3.3.6-pl2.tar.gz
$ gzip -d fftw-3.3.6-pl2.tar.gz
$ tar xvf fftw-3.3.6-pl2.tar
$ cd fftw-3.3.6-pl2Now I load the gcc and openmpi modules:
$ module load gcc/6.2.0
$ module load openmpi/2.1.0Now I configure the build:
$ ./configure --prefix=/work/ccresearch/jedicker/fftw --enable-mpi --enable-shared
Then I build everything:
$ make
If everything builds (it should), then I just do:
$ make install
This will create directories for bin, include, lib, and share in my /work/ccresearch/jedicker/fftw directory
To test this, I downloaded a simple FFTW with mpi sample program I found on via google:
$ mkdir /work/ccresearch/jedicker/fftw-test
$ cd /work/ccresearch/jedicker/fftw-test
$ wget http://micro.stanford.edu/mediawiki-1.11.0/images/Simple_mpi_example.tar
$ tar xvf Simple_mpi_example.tar
$ cd simple_mpi_exampleIn the source for the simple example, I needed to modify the Makefile accordingly:
LD_RUN_PATH=/work/ccresearch/jedicker/fftw
PRJ=simple_mpi_example
simple_mpi_example: simple_mpi_example.c
mpicc -O3 -I/${LD_RUN_PATH}/include $< -L${LD_RUN_PATH}/lib -lfftw3_mpi -lfftw3 -o $@
qsub: simple_mpi_example
qsub simple_mpi_example.pbs
watch 'ls && qstat'
clean:
rm -f *~ simple_mpi_example.o
clean_all: clean
rm -f ${PRJ} simple_mpi_example.o[1-9]* simple_mpi_example.e[1-9]* simple_mpi_example.out simple_mpi_example.log
emacs:
emacs -nw Makefile simple_mpi_example.pbs simple_mpi_example.c
tar: clean_all
tar -cvf ../${PRJ}.tar ../${PRJ}
Now, in order to use the libraries that are in /work/ccresearch/jedicker/fftw/lib, I need to set my LD_LIBRARY_PATH environment variable to use that path. I just do:
$ export LD_LIBRARY_PATH=/work/ccresearch/jedicker/fftw/lib:$LD_LIBRARY_PATH
Then I can run the program:
$ mpirun -n 4 ./simple_mpi_example
2500
2500
2500
2500
finalize
finalize
finalize
finalize
The important thing is that in order to get everything to work, I need to do this:
$ module load gcc/6.2.0
$ module load openmpi/2.1.0
$ export LD_LIBRARY_PATH=/work/ccresearch/jedicker/fftw/lib:$LD_LIBRARY_PATHYou will probably need to add something similar to your sbatch script.
How can I see html files on a cluster?
Firefox browser needs a lot of memory to run and can not be started on the clusters' head nodes due to strict limits. You can login to the appropriate DTN node, novadtn, and start browser there.
I don't want my jobs to requeue on failure, how do I stop slurm from doing that?
Jobs that requeue typically overwrite output files. There are many occasions where this is not desirable.
#SBATCH --no-requeue
In your batch script will prevent slurm from requeueing your job automatically.
How do I use GNU Parallel to maximize my usage of a node?
To get started add
module load parallel
in your job script to give you access to GNU Parallel.
The easiest way to use parallel is to create a list of bash scripts that you wish to run, with completely independent scripts, one per line, all to be run on a single node. For an example, and simplicity, assume that you want to have 150 single R commands run, each using 24 G.
Only 5 of the scripts can run at a time on a 128GB node without causing memory swapping. We could just have 5 of the R commands per script, which would fill the node, but since some of these finish earlier than others, there would be quite a bit of idle time.
We suggest placing 15 command in each of 10 scripts, and limit each to just run 5 at a time. Parallel can be invoked to run just 5 of the R command at a time, starting a new one as soon as one of the first 5 is complete, and so on until all the commands are completed, or the job runs out of time.
To do this first create scripts my_parallel_script0 , my_parallel_script1, … , my_parallel_script9. Each with 15 of your R commands, one per line.
Submit 10 jobs, each having a reservation of a full node use somethig like:
-N 1 –n 16 –mem=120g –t -t 48:00:00
The job 0 will have the command: parallel –j 5 < my_parallel_script0
job1 will have the command: parallel –j 5 < my_parallel_script1
If you want a simple example,
Put the following in a batch script b
#!/bin/bash
sleep 10
sleep 2
sleep 5
sleep 3
sleep 1
sleep 7
sleep 12
And make it executable by issuing
chmod u+x b
the command
time ./b
returns a time just over 40 seconds
while
time parallel –j 3 < b
returns in just over 18 seconds.
If you know which of the lines tales the longest, and can sort form longest to shortest, You will get the minimal speed. Doing so in the above b script and producing the c script:
#!/bin/bash
sleep 12
sleep 10
sleep 7
sleep 5
sleep 3
sleep 2
sleep 1
then
time parallel –j 3 < c
returns in just over 13 seconds. 12 seconds would be a minimum time no matter how large j is, since the longest sleep time is 12 seconds. You may want to use more commands in a single my_parallel_script or more of the parallel scripts.
This should provide you with the best of use of resources.